|
|||
| 新闻来源:江苏艾迪生生物科技有限公司 发布时间:2024.03.19 浏览次数: | |||
|
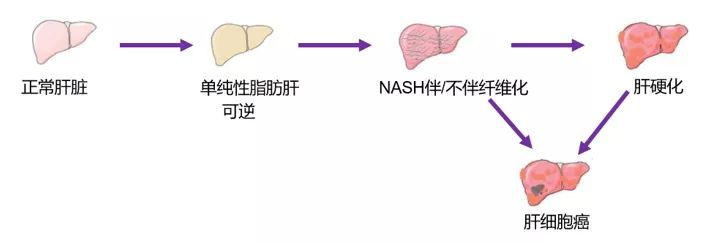
一、非酒精性脂肪性肝炎简介 非酒精性脂肪性肝炎(nonalcoholic steatohepatitis, NASH)是一种常见的肝损伤,是一种代谢紊乱症,伴随着不同程度的炎症和纤维化,最终将演变成终末期肝病(如肝硬化,肝癌),其组织病理学异常与酒精性脂肪性肝炎相似。组织病理学特征包括脂肪变性、肝细胞损伤证据、混合性炎症性小叶浸润和可变纤维化。二次打击学说是目前较为普遍接受的一种解释NASH发病机制的假说,一次打击是由肝脏中脂肪堆积造成的,脂肪变性后的肝细胞比正常肝细胞更容易受到第二次打击的损伤,导致肝细胞的一系列损伤包括炎症,凋亡,纤维化和癌变等,启动第二次打击的主要因素包括氧化应激,脂质过氧化,促炎症因子、线粒体功能障碍、肝脏微循环障碍等。
图1 非酒精性脂肪性肝炎形成过程 二、常用模型的建模方法 根据脂肪肝发病机理,可将构建原理分为以下5类: 1.通过改变高脂饲料营养结构,使动物摄入或合成过多脂肪,或对高脂饲料进行特殊处理,使动物肝脏受损,构建脂肪肝模型; 2.寻找自然发生的特定基因突变,或靶向基因修饰,诱导动物产生肥胖、肝脏脂肪变性等症状; 3.使用药物或激素,干预动物脂肪合成与代谢过程,模仿脂肪肝症状; 4.高脂饲料与基因敲除或药物多种模型混合诱导,使脂肪肝模型构造周期缩短,症状更加全面; 5.使用天然动物细胞作为脂肪紊乱模型,无需额外药物试剂诱导。 根据模型制作方法,非酒精性脂肪肝动物模型可分为饮食诱导、化学物质诱导、基因编辑等。 1饮食诱导 1.1 营养失调性脂肪肝模型 1.1.1高脂饮食(Highfatdiet,HFD)模型 (1)HFD是最基础的非酒精性脂肪肝病造模方式,此模型大致模拟了西方饮食。HFD中含有较多的脂肪合成原料,可促进肝脏脂肪生成,过度堆积脂肪,从而导致脂肪肝。 由于C57BL/6小鼠对高脂饲料比较敏感,容易形成高脂血症,故而是常用的复制NASH模型的动物。 长期喂食HFD后,动物主要会出现肥胖、IR、单纯脂肪变性等症状。常见的HFD包含71%脂肪、11%碳水化合物和18%蛋白质。 HFD炎症严重程度与处理时间成正比,目前对于非酒精性脂肪性肝炎(NASH)模型,大部分研究的HFD诱导时长为12~20周。 喂食高脂饮食的小鼠模型模拟了人类NAFLD的组织病理学和发病机制,因为它具有NAFLD患者中观察到的主要特征,包括肥胖和胰岛素抵抗。因此,该模型适于评估减重、低脂饮食及改善胰岛素抵抗的治疗药物。从发病机制、病程的发生发展及最终组织形态学改变与人类组织的差异,及动物的死亡率、成本代价上来考虑,最常采用的模型为饮食诱导型,高脂饮食模型也有一定的局限性,如造模耗时较长,至少需两个月以上,而且随着动物种系和饮食配方的不同呈现出多样性。 (2)近期也有研究表明,高脂饲料(HFD,配方为普通饲料53.5%、果糖20%、猪油20%、胆固醇5%、食盐1%、胆酸钠0.5%)联合不同浓度葡聚糖硫酸钠(DSS)饮水导致小鼠非酒精性脂肪肝炎(NASH)的动物模型,给予2%的DSS可加速高脂饮食诱导小鼠NASH模型的建立,可将造模时间缩短至2周,大大缩短造模时间。 1.1.2致动脉粥样硬化饮食(ATH)模型 ATH含有0.5%胆酸盐和1.25%的胆固醇,通常应用于小鼠以构建粥样硬化斑块模型。 研究发现,ATH可致C57BL/6小鼠出现血脂异常。与此同时,在6-24w内,ATH还引发了NASH,且造模时间越长,病理状态越严重,如出现肝细胞大泡性脂肪性变,炎症以及肝纤维化等。此外,小鼠在6w后也表现出丙氨酸氨基转移酶、总胆固醇和甘油三酯水平升高。 然而,ATH并不致体重增加,也没有明显的胰岛素抵抗现象,因此,与人类NASH病理机制有一定区别,成为该模型的缺陷。 1.1.3高脂+致动脉粥样硬化饮食(HFD+ATH)模型 HFD+ATH是在高脂饮食的基础上,添加0.5%的胆酸盐和1.25%的胆固醇。该模型结合了高脂模型及ATH模型的优点,可有效地促进非酒精性脂肪肝炎的进一步发展,主要病理特征为胰岛素抵抗、氧化应激、肝星状细胞激活。 相关研究表视将HFD+ATH(60%脂肪+1.25%胆固醇+0.5%胆酸盐)与胆碱-蛋氨酸缺乏饮食进行分析比较,两者具有类似的NASH病理特征,HFD+ATH诱导的C57BL/6小鼠表现出肝胆固醇和游离脂肪酸增加,而胆碱-蛋氨酸缺乏饮食的小鼠为肝脏的甘油三酯水平升高为主。 1.1.4高脂+胆固醇饮食模型 高脂+胆固醇饮食是在高脂饮食的基础上,添加21%的黄油和0.2%的胆固醇。 短期喂养高脂+胆固醇饮食后,C57BL/6J小鼠仅发生肝脏脂肪变性。而雌性ldlr-/-和APOE2ki小鼠会表现出严重的炎症和脂肪变性,其特征为巨噬细胞浸润和NF-kappaB介导的信号增强。雄性ldlr-/-和APOE2ki小鼠也出现严重的肝脏炎症,但未有脂肪变性。 此外,一种西方饮食,其中有富含n-6-PUFA大豆油(100g大豆油含有25g的n-6-多不饱和脂肪酸)和0.75%胆固醇。C57BL/6小鼠被给予该饮食20w后,小鼠表现出肝脂肪变性呈气球样变、氧化应激、炎性细胞浸润、Kupffer激活和肝纤维化,伴有体重增加、胰岛素抵抗的NAFLD特征。 1.1.5高脂+高糖饮食(ALIOS)模型 ALIOS是一种高脂与高糖饮食协同诱导的小鼠模型,ALIOS造模方法是高脂饲料(含30%的反式脂肪酸)及果葡糖浆喂养,16w后,C57BL/6小鼠有明显的脂肪变性,伴有坏死性炎症,丙氨酸转氨酶水平升高。 与仅高脂饲料(含30%的反式脂肪酸)喂养相比,ALIOS组动物体重明显增加,食物摄入量增加,胰岛素抵抗增强,但脂肪变性程度和丙氨酸转氨酶水平几乎无变化。 有文献报道,通过使用西方饮食(Tekladdiets,TD.120528)饲料,辅以高果糖的饮用水,两周一次腹腔注射小剂量的CCl4时,发现C57BL/6小鼠可快速地从单纯性脂肪肝到脂肪性肝炎,肝纤维和肝细胞癌的演进,几乎符合人类NAFLD的病理过程。 1.1.6高脂+高果糖+高胆固醇饮食(AMLN,GAN)模型 为了弥补先前报道的ALIOS小鼠模型的缺陷,更加客观评估动物肝脏病理进程,可通过高脂+高果糖+高胆固醇(AMLN)饮食模型,其含40%的脂肪(其中约18%的反式脂肪酸),22%的果糖和2%的胆固醇。 C57BL/6和ob/ob小鼠同时喂养AMLN(26-30w)后,均出现明显的肝脂肪变性、肝小叶炎性细胞浸润和肝细胞形态肿胀。 值得注意的是,C57BL/6小鼠需喂养26-30w后,才会出现上述病理特征。相比于C57BL/6小鼠,ob/ob小鼠喂养AMLN12w后,则可表现出甘油三酯和丙氨酸氨基转移酶水平明显升高。 另外一种含有41%的脂肪,30%的果糖和2%的胆固醇的高脂+高胆固醇+高果糖饮食可很好地模拟西方饮食中高脂高糖高胆固醇的习惯,该饮食模型适用性更为广泛,已被用于建立多种基因型小鼠的非酒精性脂肪肝炎模型。 研究报道,相关研究提出了GAN饮食模型,配方含40%的脂肪 (无反式脂肪酸),22%的果糖,10%蔗糖和2%的胆固醇。ob/ob小鼠喂养GAN8-16w后,表现出明显的体重增加,且与人类NASH的病理特征十分相似。此外,与AMLN饮食的ob/ob小鼠的病理特征也具有一致性,但GAN饮食的ob/ob小鼠的体重增加更为明显。 相比之下,C57BL/6小鼠则需要较长的喂养时间(通常需要28w后)才能诱导出与上述一致的NASH模型。 1.1.7胆碱-蛋氨酸缺乏饮食(MCD)模型 胆碱-蛋氨酸缺乏饮食含40%的蔗糖和10%的脂肪,但是不含有蛋氨酸和胆碱。最初2-4w内小鼠就会出现NAFLD病理表型的主要特征,如小鼠的肝细胞大泡性脂肪性变,并随后出现明显的纤维化。 尽管MCD模型是研究NASH的重要工具,并用于早期药物的筛选,但MCD模型也有一定的局限性,如胆碱-蛋氨酸缺乏饮食配制繁琐,不符合人类饮食规律,也与人类NASH患者的实际代谢状况完全不同,缺乏人类NASH所具有的全身性胰岛素抵抗特征。 相关研究发现MCD模型呈现出低血清胰岛素、空腹血糖、瘦素和甘油三酯水平。与NASH患者多有肥胖不同,MCD模型较正常对照组出现严重体重下降,呈恶病质状态。 1.1.8胆碱缺乏L-氨基酸饮食(CDAA)模型 CDAA饮食含有68.5%的碳水化合物、17.4%的蛋白质和14%的脂肪。CDAA饮食诱导模型与MCD小鼠模型类似,它可以抑制肝细胞的脂肪酸氧化,增加脂质合成,氧化应激和炎症,最终导致纤维化。 长期喂食CDAA的小鼠也可出现体重增加、胰岛素抵抗和血脂水平显著升高。另外,CDAA喂养小鼠出现代谢相关症状的时间长,目前仅限于肝脏疾病研究,不宜单独应用于NASH相关研究。 2、药物诱导脂肪肝模型 2.1链脲佐菌素(STZ)诱导模型 链脲佐菌素诱导模型常见于诱导2型糖尿病的实验性模型,C57BL/6小鼠在出生2d后,给予200μg/L剂量,并结合高脂饮食喂养,大约20w内出现脂肪性肝炎、纤维化甚至肝癌,可以作为NAFLD的小鼠模型。 STZ诱导模型小鼠6周龄出现单纯性肝脏脂肪变性,8w时出现小叶炎症和肝细胞广泛气球样变,8-12w时肝脏脂肪沉积向纤维化发展,20w时小鼠高发肝细胞癌。造模期间,小鼠的转氨酶和血糖浓度一直呈现升高趋势。 该模型可以诱发氧化应激、炎症、纤维化和肝细胞凋亡,符合NAFLD的组织病理学。但与人类NAFLD的胰岛素抵抗不同的是,链脲佐菌素会破坏胰岛β细胞功能,而不会导致全身性的胰岛素抵抗。 因此,这也限制该模型在临床前药物研究中的使用。 2.2四氯化碳(CCl4)诱导模型 CCl4单独诱导模型仅出现肝纤维化,而不发生肥胖和胰岛素抵抗等病变,因此不属于NAFLD模型。 因此,CCl4通常需要与饮食诱导模型相结合制备NASH模型,如CCl4可以促进高脂饮食对非酒精性脂肪肝炎和肝纤维化的发展。 3基因编辑诱导模型 3.1 ob/ob和db/db 肥胖基因(OB)所表达的瘦素通过影响新陈代谢和食欲来调节体重和脂肪沉积,其含量与脂肪沉积程度成反比,瘦素受体是由糖尿病相关基因(db基因)编码产生的蛋白质,DB基因所编码的瘦素受体缺失容易引起高血糖症或IR。 ob/ob或db/db小鼠先存在隐性突变,先天缺少显性OB或DB基因,无法编码瘦素或瘦素受体,其特征是肝脏脂质积累增多,不自发产生炎症和纤维化,表现出脂肪肝的明显代谢特征,是脂肪肝的良好遗传模型,两者不同之处在于,ob/ob小鼠可以抵抗肝纤维化,而db/db小鼠却不抵抗肝纤维化。 然而,先天性瘦素缺乏或基因突变引起的瘦素抵抗在肥胖或NASH患者中并不普遍,因此ob/ob和db/db小鼠模型在反映动物肥胖或NASH的成因方面能力有限。 3.2Dicer1基因敲除 为了开发与人类非酒精性脂肪性肝炎(Non-alcoholic steatohepatitis,NASH)特征相似的动物疾病模型,本试验利用肝脏特异性Dicer1基因敲除(KO)小鼠,经高脂高糖饮食诱导12周构建了一种新型NASH小鼠模型。 3.3 129S1/SvImJ 和 C57BL/6J杂交小鼠 使用两种常见小鼠品系 129S1/SvImJ 和 C57BL/6J 杂交的同基因菌株诱导非酒精性脂肪肝模型,其中简单的高脂肪饮食伴随着随意消耗果糖和葡萄糖含量高的水(西方饮食糖水 (WD SW)),目前已经在两种小鼠品系(129S1 / SvImJ和C57Bl / 6J)之间开发了非酒精性脂肪性肝炎(NASH)和肝癌的饮食诱导小鼠模型。该模型模拟了人类NASH的所有生理,代谢,组织学,转录组学基因特征和临床终点,并且可以促进NASH治疗靶点的临床前开发。 4复合模型 基因编辑模型无法独立发展为脂肪肝,因此许多动物模型结合自然发生的基因突变或靶向基因修饰与日粮诱导两种方法,使模型的组织病理学和病理生理学更接近于动物脂肪肝。 例如: (1)雌性Alms11突变体(foz / foz)小鼠喂食致动脉粥样硬化饮食(23%脂肪,45%碳水化合物,20%蛋白质,0.2%胆固醇)持续16周,foz/foz小鼠体重>60g,表现为高胰岛素血症、糖尿病、高血压、高胆固醇血症、低脂联素血症和NASH伴纤维化,且此混合模型更能模仿人类的脂肪肝状况[7]。 (2)通过给8~12周龄ob/ob和db/db小鼠饲喂MCD日粮(10%脂肪、40%蔗糖、缺乏蛋氨酸和胆碱),4周后观察到肝脏中的脂肪浸润严重,硫代巴比妥酸反应物水平和亚硝酸盐的增加,且db/db小鼠比ob/ob和野生型小鼠有明显更高的血清ALT水平和更严重的肝脏炎症和纤维化。由于肝组织的脂肪变性和氧化应激密切相关,这些变化可能促进了动物的NASH。 也有研究对ddb/db小鼠饲喂高铁元素日粮,观察到日粮中的铁超载可以加速动物脂肪肝向NASH的发展进程。 (3)有研究对30周龄OLETF大鼠饲喂含60%果糖的HF饮食,发现其产生了血脂异常和炎症等症状,BiP、p-IRE1α和t-IRE1α蛋白表达量显著增加,表明内质网应激通路被激活。 研究者应根据实验研究目的,选择符合人类脂肪肝形成过程及病变特征、造模时间短、动物死亡率低、重复性好、操作方法较简便的动物模型进行研究。 三、目前上市药物 目前针对NASH几乎无药可治,患者有着巨大的未满足临床需求,迫切需要开发有效的治疗方法来解决这一全球健康问题。NASH是新药研发的热门领域,近年来投入研究的新药层出不穷。目前用于NASH治疗的药物包括核受体激动剂,如FXR激动剂、PPAR激动剂、趋化因子受体抑制剂、甲状腺激素受体-β激动剂以及GLP-1、FGF 21或SGLT2抑制剂等。 PPAR受体是一种调节代谢稳态、细胞分化和免疫炎症的核受体,也是近年来比较热门的NASH治疗靶点。一种PPARα/γ双重激动剂saroglitazar已在印度率先批准上市,2020年3月,印度药品管理局批准Saroglitazar用于NASH的治疗,这也是世界上第一个获批用于治疗非肝硬化性NASH的药物,但该适应证尚未通过美国食品药品管理局(FDA)和欧盟药品管理局的审批,该药在国际上尚未被广泛接受。 四、作用靶点
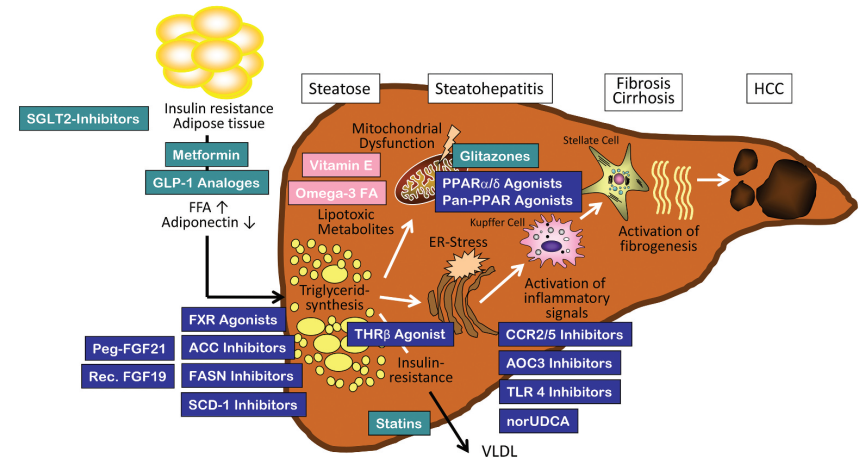
图2. NASH的发病机制和新的治疗靶点 1. PPAR受体 PPAR受体是一种调节代谢稳态、细胞分化和免疫炎症的核受体,也是近年来比较热门的NASH治疗靶点。 2. 甲状腺激素β受体 甲状腺激素β受体是目前比较被看好的治疗靶点。甲状腺激素受体β激动剂主要通过促进肝脏脂代谢和降低脂毒性来改善NASH, 其中MGL-3196和VK2809已进入后期临床阶段,并且表现出了较为显著的降低肝脂肪含量的效果。 3. SCD-1 硬脂酰辅酶A去饱和酶1(SCD-1)是一种在肝脂肪生成过程中发挥作用的关键酶,可将饱和脂肪酸转化为单不饱和脂肪酸。近年来,SCD-1已成为NAFLD饮食代谢模型的主要研究靶点。 4. XBP1 XBP1是一种转录因子,在非酒精性脂肪性肝炎(NASH)患者的肝组织中上调。肝细胞中Xbp1的条件敲除导致小鼠脂质积累减少,而巨噬细胞中Xbp1的遗传缺失改善了小鼠的营养性脂肪性肝炎和纤维化。XBP1的药理学抑制可预防脂肪性肝炎和纤维化。 5. ALOX12- ACC1轴 有研究者在多组学研究和跨物种多层验证的基础上,表明花生四烯酸12-脂氧合酶(ALOX12)-乙酰辅酶A(CoA)羧化酶1(ACC1)轴是NASH进展的核心驱动因素已有研究表明,ALOX12参与肝损伤,如肝缺血再灌注损伤和Ccl4引发的肝损伤,ALOX12的酶抑制剂ML355可以有效改善血栓形成,血小板聚集和胰岛功能障碍。 虽然ALOX12主要通过其产生12-羟基二十碳四烯酸(12-HETE)的酶活性参与上述研究,但我们的配套研究表明,ALOX12通过酶活性非依赖性方式促进NASH进展。以前的报告已经明确了ACC作为NASH的一个有吸引力的治疗靶点,因为在实验观察和临床试验中,ACC抑制剂对肝脂肪变性和纤维化的有力改善。 五、参考文献 [1] Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149: 389–397e310. [2]Paradies G, Paradies V,Ruggiero FM,et al.Oxidative stress,cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease[J].World J Gastroenterol, 2014,20(39):14205-14218. [3]Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol. 2018 Feb;68(2):230-237. doi: 10.1016/j.jhep.2017.10.031. Epub 2017 Nov 9. PMID: 29128391; PMCID: PMC5775040. [4]万小雨,曾俊豪,张婉怡,韩钘雨,杨天心,王雅琴,周永芹,刘朝奇.非酒精性脂肪性肝炎小鼠模型的建立[J].中国免疫学杂志,2020,36(16):1925-1930. [5]苏晓鸥,陈静,戚新明,周向梅,乔俊文,任进.新型非酒精性脂肪性肝炎小鼠模型的建立[J].中国兽医杂志,2019,55(08):87-91+95+132. [6]Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min HK, Mirshahi F, Bedossa P, Sun X, Hoshida Y, Koduru SV, Contaifer D Jr, Warncke UO, Wijesinghe DS, Sanyal AJ. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol. 2016 Sep;65(3):579-88. [7] Larter CZ, Yeh MM, Van Rooyen DM, Teoh NC, Brooling J, Hou JY, et al. Roles of adipose restriction and metabolic factors in progression of steatosis to steatohepatitis in obese, diabetic mice. J Gastroenterol Hepatol 2009;24: 1658–1668. [8] Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, et al. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011;141: 1393–1403. [8]RATZIU V, HARRISON SA, FRANCQUE S, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and-δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening[J]. Gastroenterology, 2016, 150(5): 1147-1159. e5. [9] SHARMA M, PREMKUMAR M, KULKARNI AV, et al. Drugs for non-alcoholic steatohepatitis (NASH): Quest for the holy grail[J]. J Clin Transl Hepatol, 2021, 9(1): 40-50. [10]Monika Rau, Andreas Geier. An update on drug development for the treatment of nonalcoholic fatty liver disease – from ongoing clinical trials to future therapy [J]. Expert Review of Clinical Pharmacology. 2021, 14(3): 333-340. [11] RATZIU V, HARRISON SA, FRANCQUE S, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and-δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening[J]. Gastroenterology, 2016, 150(5): 1147-1159. e5. [12] ALKHOURI N. Thyromimetics as emerging therapeutic agents for nonalcoholic steatohepatitis: Rationale for the development of resmetirom (MGL-3196)[J]. Expert Opin Investig Drugs, 2020, 29(2): 99-101. [13] SAFADI R, KONIKOFF FM, MAHAMID M, et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease[J]. Clin Gastroenterol Hepatol, 2014, 12(12): 2085-2091. e1. [14]Wang Q, Zhou H, Bu Q, Wei S, Li L, Zhou J, Zhou S, Su W, Liu M, Liu Z, Wang M, Lu L. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J Hepatol. 2022 Aug;77(2):312-325. [15]X. J. Zhang, Z. G. She, J. Wang, D. Sun, L. J. Shen, H. Xiang, X. Cheng, Y. X. Ji, Y. P. Huang, P. L. Li, X. Yang, Y. Cheng, J. P. Ma, H. P. Wang, Y. Hu, F. Hu, S. Tian, H. Tian, P. Zhang, G.-N. Zhao, L. Wang, M. L. Hu, Q. Yang, L. H. Zhu, J. Cai, J. Yang, X. Zhang, X. Ma, Q. Xu, R. M. Touyz, P. P. Liu, R. Loomba, Y. Wang, H. Li, Multiple omics study identifies an interspecies conserved driver for nonalcoholic steatohepatitis. Sci. Transl. Med. 13, eabg8117 (2021). [16]C. Dai, H. Li, Y. Wang, S. Tang, T. Velkov, J. Shen, Inhibition of oxidative stress and ALOX12 and NF-κB pathways contribute to the protective effect of baicalein on carbon tetrachloride-induced acute liver injury. Antioxidants 10, 976 (2021). [17]F. Yang, Y. Zhang, H. Ren, J. Wang, L. Shang, Y. Liu, W. Zhu, X. Shi, Ischemia reperfusion injury promotes recurrence of hepatocellular carcinoma in fatty liver via ALOX12-12HETE-GPR31 signaling axis. J. Exp. Clin. Cancer Res. 38, 489 (2019). [18]X. J. Zhang, Z. G. She, J. Wang, D. Sun, L. J. Shen, H. Xiang, X. Cheng, Y. X. Ji, Y. P. Huang, P. L. Li, X. Yang, Y. Cheng, J. P. Ma, H. P. Wang, Y. Hu, F. Hu, S. Tian, H. Tian, P. Zhang, G.-N. Zhao, L. Wang, M. L. Hu, Q. Yang, L. H. Zhu, J. Cai, J. Yang, X. Zhang, X. Ma, Q. Xu, R. M. Touyz, P. P. Liu, R. Loomba, Y. Wang, H. Li, Multiple omics study identifies an interspecies conserved driver for nonalcoholic steatohepatitis. Sci. Transl. Med. 13, eabg8117 (2021) [19]C. Dai, H. Li, Y. Wang, S. Tang, T. Velkov, J. Shen, Inhibition of oxidative stress and ALOX12 and NF-κB pathways contribute to the protective effect of baicalein on carbon tetrachloride-induced acute liver injury. Antioxidants 10, 976 (2021). [20]H. He, R. Adili, L. Liu, K. Hong, M. Holinstat, A. Schwendeman, Synthetic high-density lipoproteins loaded with an antiplatelet drug for efficient inhibition of thrombosis in mice. Sci. Adv. 6, eabd0130 (2020). |
|||
| 本文共分 1 页 | |||
|